

药学院范骁辉教授团队开发注释单细胞转录组测序数据细胞类型的新工具scDeepSort
2021年9月9日,浙江大学药学院范骁辉教授与计算机学院陈华钧教授团队在Nucleic Acids Research在线报道了最新研究成果—“一种基于加权图神经网络的深度学习方法”(scDeepSort: a pre-trained cell-type annotation method for single-cell transcriptomics using deep learning with a weighted graph neural network),用于自动注释单细胞转录组测序(single-cell RNA sequencing,scRNA-seq)数据的细胞类型(图1)。研究团队将细胞与基因作为图神经网络的节点,对已知细胞类型的scRNA-seq数据进行有监督学习,从而实现对新数据集的预测,为准确注释scRNA-seq数据的细胞类型提供了新的解决方案。
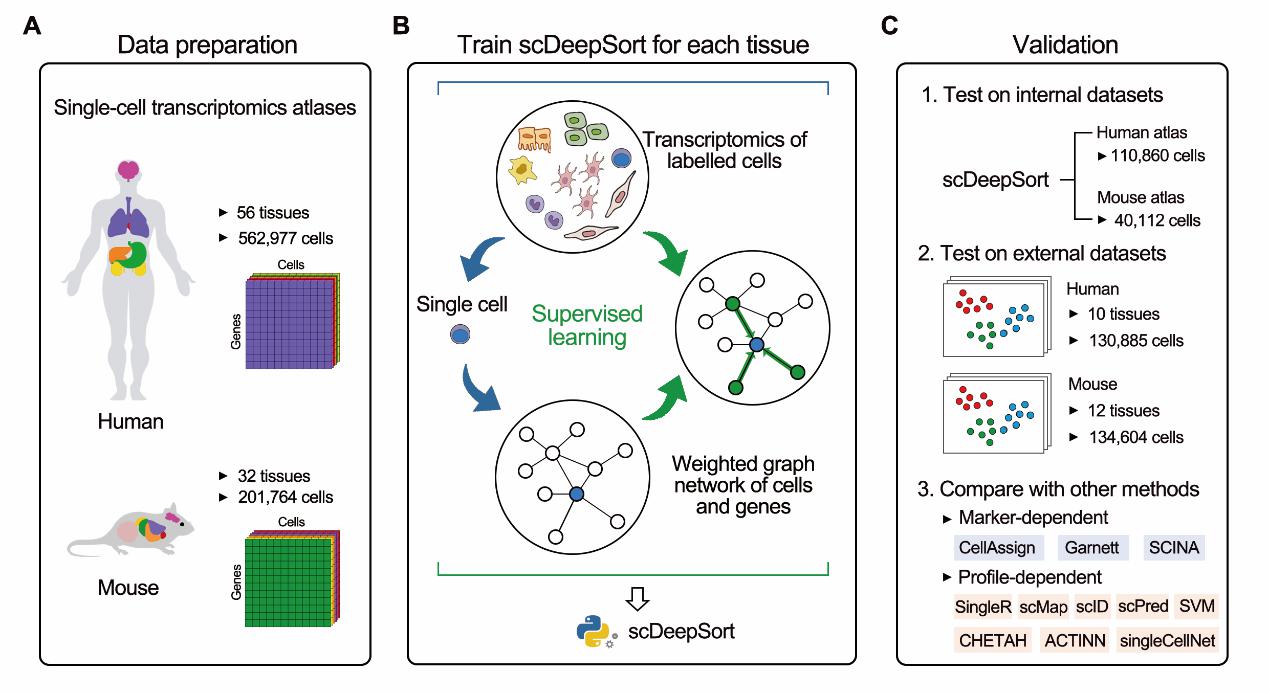
图1. scDeepSort的工作流程
细胞类型注释是scRNA-seq数据分析流程中的首要步骤,在scRNA-seq数据的谱系追踪、细胞间通讯推断中都需要先确定每个细胞的类型/亚型,方可进一步分析。目前方法主要是通过将单细胞与已知细胞类型的参考数据(标记基因或表达谱)进行相似性比较,从而确定单细胞的细胞类型。然而,大部分方法非常依赖用户提供的参考数据,并且注释的准确率不高。在此项研究中,研究人员尝试以GraphSAGE为框架,以细胞和基因为节点构建图网络,充分利用图结构的信息,通过加权聚合使得信息在图网络中充分传播,并产生线性可分离的特征空间,最后通过概率将加权图聚合器层产生的细胞表示判定为预定义的细胞类型(图2)。通过加权图神经网络的预训练,scDeepSort可准确预测不同组织的单细胞类型。与现有的其他方法相比,scDeepSort在76个测试集(265,489个细胞)上的准确率达83.78%,排名第一。
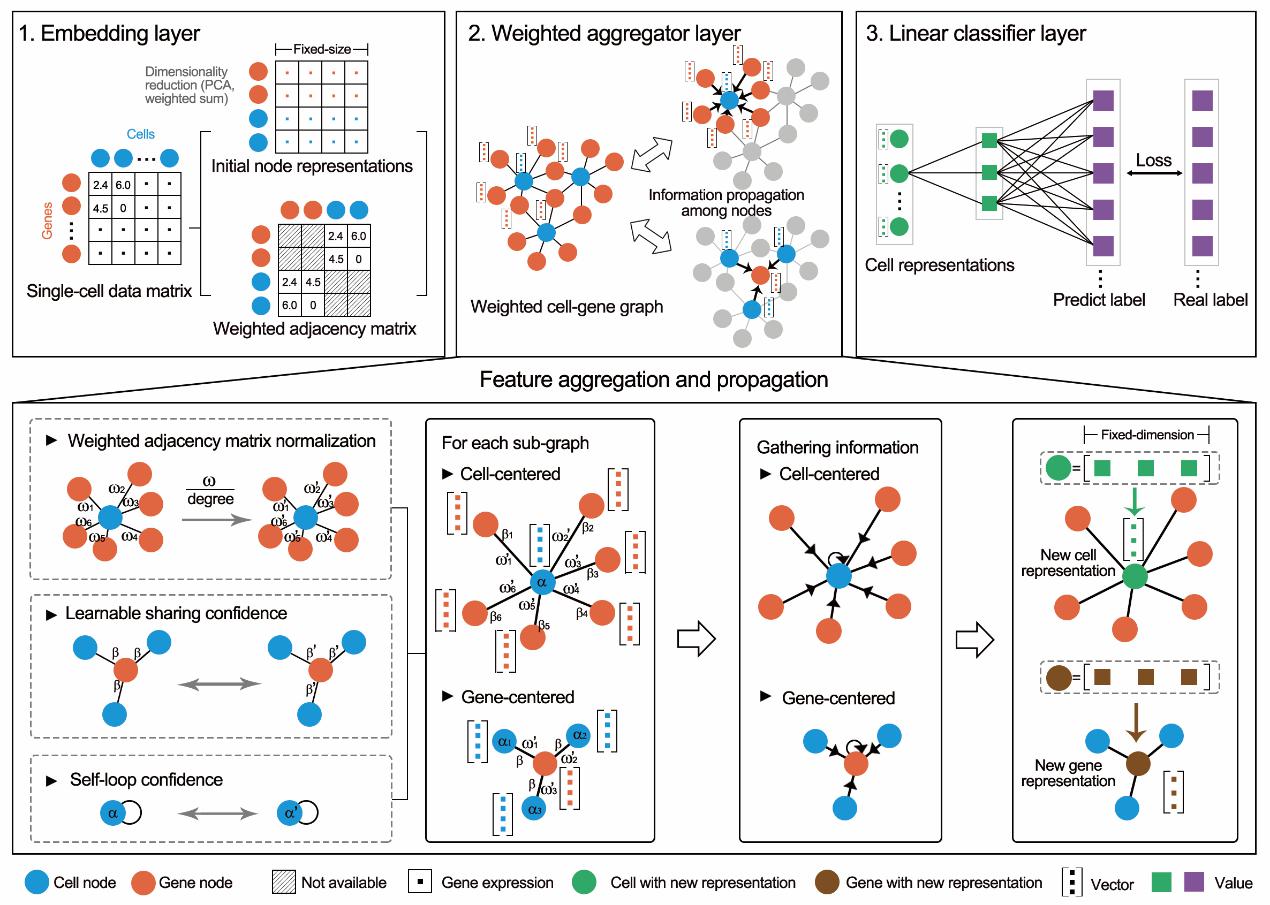
图2. scDeepSort的算法示意图
单细胞测序是近年发展起来的新兴技术,在药学和中医药研究领域备受瞩目,但还存在着大量的技术难题和挑战。范骁辉教授团队与合作者通过技术创新在该领域做了大量尝试,相关成果在Trends in Biotechnology (2021)、Briefings in Bioinformatics (2021)、Computational and Structural Biotechnology Journal (2021)、Protein & Cell (2020)、iScience (2020)等期刊发表。
本研究受到国家自然科学基金、浙江省自然科学重点基金和国家高层次青年人才等项目资助,并得到了阿里云的支持。本文第一作者为浙江大学药学院博士后邵鑫和计算机学院2017级直博生杨海宏,通讯作者为浙江大学药学院范骁辉教授和计算机学院陈华钧教授。
药闻热点
21
2021.09
14
2021.09
14
2021.09
13
2021.09
13
2021.09
10
2021.09
09
2021.09
18
2021.08

